2020-06-12
在已知的6 000多种遗传病中, 目前只有大约5%的疾病可以通过小分子药物或酶替代疗法进行治疗,超过95%的遗传疾病无有效的治疗方案, 治愈更是无从谈起。基因治疗为遗传疾病带来了希望,尤其对于单基因罕见病,基因治疗提供了一种从根本上治愈遗传疾病的可能,即仅需一次治疗就可以达到终身治愈的目的。近几年来基因编辑技术的快速发展,迎来了一个全新的时代,以CRISPR/Cas9、单碱基编辑、CART等技术引领的产业变革越来越烈,让我们看到这些新兴技术的巨大潜力,并激发了无限的机遇。
邦耀生物自2013年成立以来,一直聚焦于基因编辑和基因治疗领域,经过几年发展,基于基因编辑和基因治疗的技术正在不断取得各种突破并逐步走向成熟。特别是近1年来,邦耀已在国际著名学术期刊Nature Biotechnology、Nature Medicine、Nature Cell Biology、Cell Research等发表多篇学术论文,特别是在单碱基编辑工具开发领域的相继突破,为基础研究和遗传性疾病如β-地中海贫血的治疗提供了新的发展方向和工具。
目前邦耀已自主研发并成功搭建起基因治疗技术转化平台,并且在基因编辑和基因治疗领域已经递交30余项全球及中国专利申请。其中,基因治疗β-地中海贫血的产品管线,邦耀生物采用了具有自主知识产权的基因编辑技术重新开启胎儿期的γ珠蛋白的表达,代替有缺陷的β珠蛋白的治疗方案,可以有效缓解地贫症状,是一种更为高效、便捷、安全且成本极低的治疗手段,具有很大的临床转化潜力。该项目正与国内多家顶级医疗单位携手,希望借此项研究及其临床实验的推广,使得基于基因编辑的基因治疗成为我国β-地中海贫血患者全新的临床治疗方案,并有望实现“一次治疗终身治愈”的疗效。
值得一提的是,除了重点布局在地中海贫血的产品管线,邦耀也正在积极拓展其他遗传疾病领域。2020年5月,邦耀生物科学家刘明耀教授及李大力教授团队,先后在基因治疗和肾脏疾病顶级杂志Molecular Therapy和Kidney International发文,表明基因治疗在I型酪氨酸血症、I型原发性高草酸尿症等遗传疾病中的巨大潜力。

2020年5月7日,基因治疗顶级杂志Molecular Therapy在线发表了题为“Amelioration of an Inherited Metabolic Liver Disease through Creation of a De Novo Start Codon by Cytidine Base Editing”的研究论文。该研究利用重组腺相关病毒(rAAV)递送拆分型胞嘧啶单碱基编辑工具BE4max,成功治愈了Ⅰ型酪氨酸血症(HTⅠ)小鼠。该研究首次开创性地利用单碱基编辑技术在小鼠基因组上引入功能性元件来治疗遗传疾病,通过单碱基编辑在起始密码子突变的延胡索酰乙酰乙酸水解酶(Fah)基因的5’UTR引入一个全新的起始密码子,重新开启该基因的表达,成功治愈了因缺失该酶而出现严重肝损伤的HTⅠ小鼠。华东师范大学杨磊,王立人和霍雅楠为本文共同第一作者。
I型酪氨酸血症
Ⅰ型酪氨酸血症(HTⅠ)是一种罕见的常染色体隐性遗传疾病,由于缺失酪氨酸代谢途径中最后一步反应所需的延胡索酰乙酰乙酸水解酶(FAH),使得酪氨酸代谢受阻,导致延胡索酰乙酰乙酸(Fumarylacetoacetate,FAA)、马来酰乙酰乙酸(Maleylacetoacetate,MAA)和琥珀酰丙酮(Succinylacetone,SA)等有毒代谢产物的积累,造成严重的肝肾损伤。目前HTⅠ的发病率为1/12000到1/100000之间, 若不给予任何治疗,患儿通常在一岁内就会死亡。
现有疗法的缺陷:
目前,服用羟基苯丙酮酸双加氧酶(4-hydroxyphenylpyruvate dioxygenase)的抑制剂尼替西农(NTBC)是临床上最有效的治疗方法。虽然NTBC为HTⅠ 病人提供了有效的治疗,但有研究表明TNBC会影响儿童的智商,学习和社会认知能力。
本技术方案优势:
ClinVar 数据显示,人类约58%的遗传疾病是由点突变引起的,需要通过碱基转换来进行修复,而HTⅠ几乎全是由点突变导致,单碱基编辑技术的出现为治疗HTⅠ和其他由点突变引起的遗传疾病提供了一个全新的策略。HTⅠ是一个理想的基因治疗模型,被修复的肝细胞具有强大的生长优势,因此,只需要修复一小部分肝细胞,在不给予NBCT治疗的情况下,这些肝细胞就会不断扩增,最终形成一个全新的健康的肝脏。
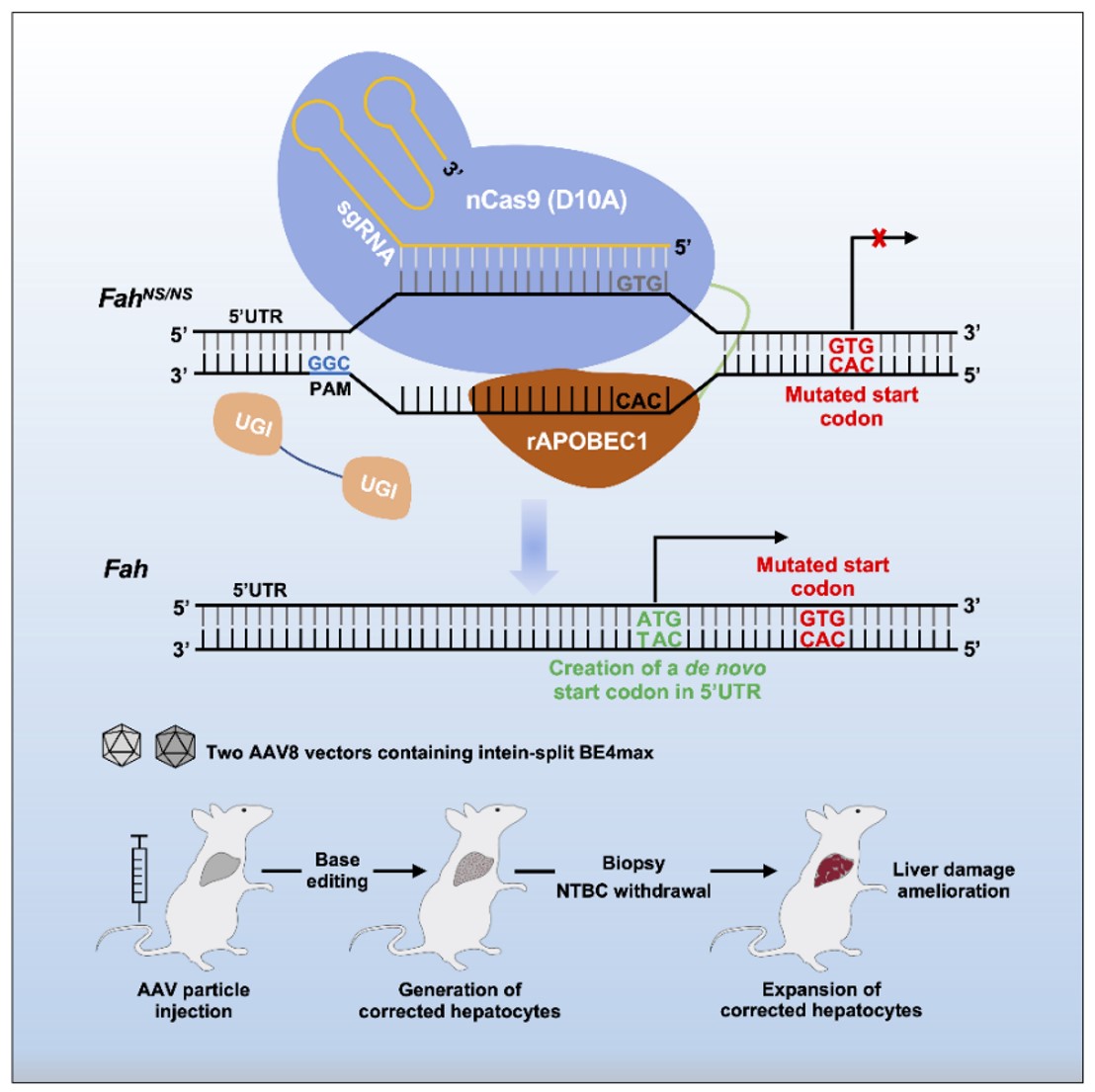
图1:治疗策略示意图
在体外实验中,研究人员利用胞嘧啶单碱基编辑工具BE4max靶向HTⅠ小鼠(FahNS/NS)Fah基因突变的起始密码子(G1TG3),但由于G1和G3都位于BE4max的编辑窗口内,导致G1和G3同时被编辑,不能有效形成正常的起始密码子(A1TG3)。为了解决这一问题,研究者另辟蹊径,利用BE4max靶向Fah基因的5’UTR区域,试图将距突变的起始密码子上游12bp处的G-12TG-10转变为一个全新的起始密码子(A-12TG-10),在该位点中,G-12位于BE4max的编辑窗口内,但G-10在编辑窗口外,经编辑可以高效地形成起始密码子(图1)。采用这种策略,研究人员利用rAAV递送拆分型BE4max到FahNS/NS小鼠肝脏,在不撤去NTBC药水的情况下,编辑效率可高达14%,撤药治疗两个月后编辑效率可高达40%,同时肝损伤得到显著缓解(图2)。
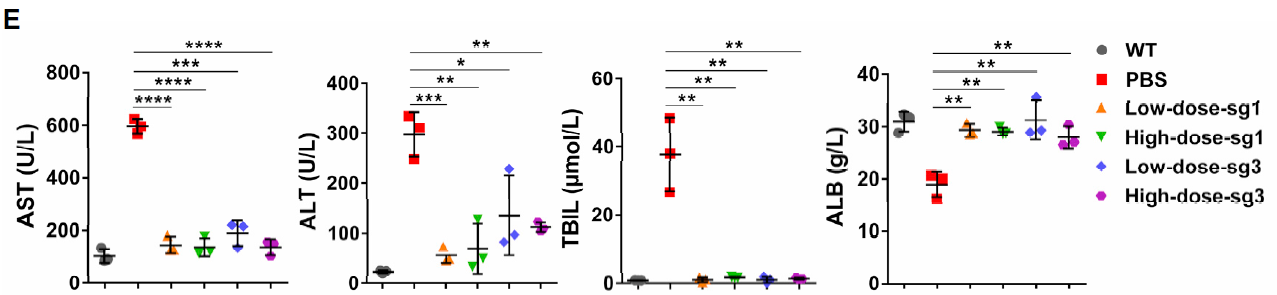
图2:血清肝功能指标检测
通过检测预测的10个脱靶位点,没有发现脱靶编辑。该研究表明单碱基编辑工具可以通过rAAV递送到肝脏并进行高效编辑,同时,通过单独引入一个起始密码子可以用于治疗由起始密码子突变而导致的遗传疾病。
二、基因疗法可以有效缓解Ⅰ型原发性高草酸尿症

2020年5月25日,华东师范大学李大力教授团队和上海交通大学医学院附属新华医院耿红全团队合作在Kidney International发文,利用CRISPR/Cas9基因编辑技术对Ⅰ型原发性高草酸尿症(PH1)大鼠进行代谢通路重编程,通过阻止上游Hao1基因的表达有效降低尿草酸水平,长期有效地缓解疾病表征。值得一提的是,该研究报道的动物模型是一种新的PH1大鼠模型, 在部分人源化的AGXT基因中携带D205N突变,出生后不久即表现出明显的高草酸和结晶尿等疾病表征,相比原有的小鼠与人类PH1疾病表现更加接近。上海交通大学医学院附属新华医院郑锐,李约延和华东师范大学王立人为本文共同第一作者。
Ⅰ型原发性高草酸尿症(PH1)
I型原发性高草酸尿症是常染色体隐性遗传疾病,由于患者体内编码丙氨酸/乙醛酸转氨酶(AGT)的AGXT基因突变所致的肝细胞内代谢异常。正常AGXT基因编码的AGT蛋白负责将乙醛酸催化成甘氨酸,AGXT的突变导致患者肝细胞内的乙醛酸无法被正常代谢,致使大量堆积的乙醛酸被氧化成草酸进入血液循环。高浓度的草酸导致肾脏和尿路结石,造成泌尿系统梗阻、肾脏损伤和终末期肾病(ESRD),超过50%的PH1患者在30岁以前进展为终末期肾病。
现有疗法的缺陷:
该病目前无特效药,保守治疗效果极差,虽然肝肾联合移植可以根治PH1,但器官供体缺乏、终身服用抗排异药物、手术创伤极大等因素使得绝大多数PH1患者无法得到根治性治疗。先后有研究团队在PH1小鼠模型(Agxt-/-)中注射载有AGXT基因的腺相关病毒(AAV)或腺病毒(Adv)进行基因治疗,草酸水平在短期内显著下降,但随着患者肝脏细胞的分裂、更新,最初递送的外源AGXT基因会逐渐丢失,治疗效果也会随之下降。需要短时间内重复治疗。另有研究显示通过SiRNA沉默乙醛酸的上游反应酶体乙醇酸氧化酶(由HAO1基因编码)可以有效切断PH1小鼠体内的乙醛酸来源进而降低小鼠体内草酸水平,但该疗法同样需要重复给药。
本技术方案优势:
相比siRNA技术和基因替代疗法,CRISPR/Cas9技术能够更高效持久地敲除体内的HAO1基因,免去了患者长期反复用药的负担。首先通过CRISPR/Cas9系统产生一个新的PH1大鼠模型,该模型在部分人源化的Agxt基因中携带D205N突变。AgxtD205N突变大鼠AGT蛋白表达缺失,在乙二醇负荷后1个月内出现高草酸尿,肾脏草酸钙(CaOx)沉积严重,提示新模型相比原有的小鼠模型与人类疾病的相关性更强。
为了验证该模型是否可用于创新疗法的开发,研究人员通过AAV载体将靶向乙醇酸氧化酶的CRISPR/Cas9系统递送到新生PH1大鼠体内。结果显示该策略在肝脏的HAO1基因中产生了30%的编辑效率,在12个月的研究周期内,治疗组的尿草酸水平比对照组降低了42%,并防止了PH1大鼠形成严重的肾草酸钙沉积。以上结果表明,这种部分人源化的AgxtD205N大鼠不仅是了解PH1病理机制的合适模型,而且能用于更多创新疗法的开发(例如文中所展示的通过基因编辑重新编程代谢途径)。
基因治疗一直被认为是最有可能从根本上治愈遗传疾病的解决方案,根据2019年发布的报告,预计在未来六年内,将有大约60种基因疗法产品获批,这些药物的销售额在2024年将达到146亿美元,约占全球医药总收入的1.2%,可见未来基因治疗罕见病的广泛应用前景。
邦耀生物自成立以来,一直放眼全球,全面布局基因治疗领域,除了重点布局基因治疗β-地中海贫血症之外,同时也重点关注酪氨酸血症、血友病等多种罕见遗传病,未来邦耀将持续通过掌握核心技术以及生产能力来保证在这一赛道的竞争力,如不断优化新型基因治疗技术(编辑工具、治疗基因的改造),及新型基因递送载体的组合治疗,早日为遗传疾病患者带来福音。
附原文链接:
1. Yang L, Wang L, Huo Y, et al. Amelioration of an Inherited Metabolic Liver Disease through Creation of a De Novo Start Codon by Cytidine Base Editing [published online ahead of print, 2020 May 7]. Mol Ther. 2020;S1525-0016(20)30235-5. doi:10.1016/j.ymthe.2020.05.001
2. Zheng R, Li Y, Wang L, et al. CRISPR/Cas9-mediated metabolic pathway reprogramming in a novel humanized rat model ameliorates primary hyperoxaluria type 1 [published online ahead of print, 2020 May 25]. Kidney Int. 2020;S0085-2538(20)30554-8. doi:10.1016/j.kint.2020.04.049

